RNA pipeline tutorial¶
This tutorial walks through the scRNA-seq branch from raw counts to the sample-level embedding. Every step shows the call you would make from a notebook, the files it writes, and the figure you should expect. Parameter values follow the shipped demo config (generate it with sampledisco --init-config).
The pipeline ends at the sample embedding. Everything after that (sample distance, trajectory, differential genes, clustering, ...) is a downstream task — see Downstream analysis.
Imports
The code below assumes sampledisco is installed (pip install sampledisco). Public functions are re-exported from each subpackage's __init__ (you can also import them from their concrete module files). The CPU implementations are shown here; GPU variants live alongside them (e.g. from sampledisco.preparation.rna_preprocess_gpu import preprocess_gpu). The config-driven wrapper switches to them automatically when RAPIDS is importable — but note the *_gpu preprocessing modules import RAPIDS at module load, so they can only be imported on a GPU box (there's no CPU fallback for those; only compute_sample_embedding has one).
Config-driven alternative
The steps below call each function directly. To run this whole branch end to end from a single YAML instead, generate the ready-to-run demo config and pass it (see the Configuration guide):
Inputs¶
RNA.h5ad— cell-level raw counts;.obsmust carry a sample column (default"sample").- (optional)
sample_meta.csv— one row per sample keyed bysample, with phenotype columns such assev.level,age,batch. Not needed when those columns already live in.obs.
Demo data
This tutorial runs on test_RNA.h5ad from the demo dataset. Download it into a local data/ folder and the snippets below work as-is — its .obs already carries sample, batch, and sev.level, so sample_meta_path can stay None.
Output lands under output_dir/rna/.
1. Preprocessing¶
Read counts, merge metadata, QC-filter cells and genes, select HVGs, compute PCA, and run a two-pass Harmony integration. A single AnnData comes out carrying two cell-level embeddings: obsm['Z_clust'] (sample-removed, used for clustering and composition blocks) and obsm['Z_rmd'] (sample-preserved, used by the RMD displacement block). Normalized expression is kept in .X, raw counts in .layers['counts'], and the HVG flag in .var['highly_variable'] (no subsetting).
from sampledisco.preparation.rna_preprocess_cpu import preprocess
adata = preprocess(
h5ad_path="data/test_RNA.h5ad",
sample_meta_path=None, # demo metadata already lives in .obs
output_dir="sampledisco_demo_output/rna",
sample_column="sample",
cell_level_batch_key=None,
min_cells=500,
min_genes=500,
pct_mito_cutoff=20,
num_cell_hvgs=2000,
cell_embedding_num_PCs=20,
num_harmony_iterations=30,
verbose=True,
)
Writes → sampledisco_demo_output/rna/preprocess/adata_preprocessed.h5ad.
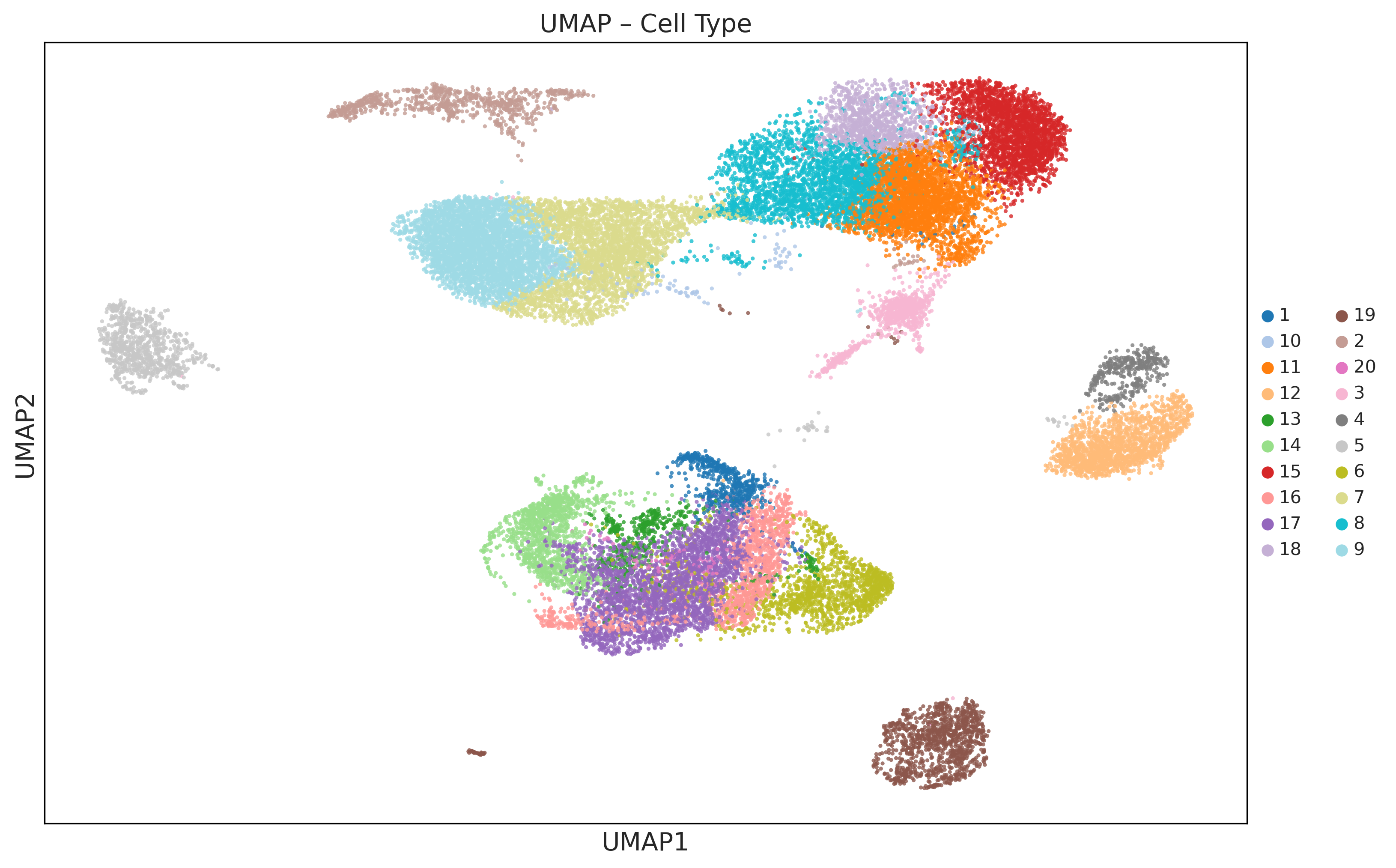
2. Cell-type clustering¶
Leiden clustering on the sample-removed Z_clust embedding, with optional UMAP and an adaptive resolution sweep to hit n_target_clusters when requested. The resulting labels are written to adata.obs["cell_type"], and the labeled cell-level AnnData is returned.
from sampledisco.preparation.cell_type_cpu import cell_types
adata = cell_types(
anndata_cell=adata,
cell_type_column="cell_type",
leiden_cluster_resolution=0.99,
n_target_clusters=None,
umap=True,
save=True,
output_dir="sampledisco_demo_output/rna",
verbose=True,
)
Writes → updated h5ad file and a UMAP PNG under preprocess/.
3. Sample embedding¶
Lift the cell-level embedding into a single sample-level embedding. SampleDisco combines multi-resolution cell-type composition blocks computed on Z_clust (coarse, medium, fine cellular states) with an RMD displacement block on Z_rmd (within-cell-type state shifts relative to a leave-one-out reference). The blocks are inverse-variance weighted, Frobenius-stacked, PCA-reduced, and Harmony-corrected at the sample level.
The result is one key, adata.uns['X_DR_sample'] — a pandas DataFrame of units × PCs (units = samples) — written in place. The function returns the modified AnnData.
from sampledisco.sample_embedding import compute_sample_embedding
adata = compute_sample_embedding(
adata,
output_dir="sampledisco_demo_output/rna",
sample_col="sample",
celltype_col="cell_type",
cluster_emb_key="Z_clust",
rmd_emb_key=None, # defaults to Z_rmd
batch_col=None,
pca_components=10,
use_rmd=True,
rmd_weight=0.60,
use_gpu=False, # CPU default; set True for RAPIDS on Linux+NVIDIA (auto-falls back to CPU)
save=True,
)
Writes → the embedding into adata.uns['X_DR_sample'], plus the embedding artifacts under output_dir.
From here, everything else — sample distance, CCA / TSCAN trajectory, trajectory DGE, sample clustering, proportion test, RAISIN cluster DGE, and visualization — runs off adata.uns['X_DR_sample'] (the downstream consumers materialize a one-row-per-sample AnnData from it via build_sample_adata). Continue to the Downstream analysis tutorials.
Tune the embedding with autotune
The RMD-vs-composition blend (rmd_weight / α) can be selected automatically with autotune (run_autotune from sampledisco.parameter_selection.autotune), enabled via the rna_autotune_enable flag on the config-driven wrapper.